微生物利用におけるリスク評価に関する技術情報の提供
微生物の安全性評価においては、対象微生物が既知有害菌に該当するかどうかの確認を行う必要があります。既知の有害菌については、種レベルでの区分でその危険度に応じたバイオセーフティレベル(BSL)が設定されており、微生物の危険性を判断する一つの指標となっています。BSLの考え方と同様に、改正感染症法における特定病原体の指定、カルタヘナ法に基づく遺伝子組換え体の第二種使用における使用区分、家畜伝染病予防法における届出病原体の指定、植物防疫法における検疫有害動植物の指定等においても、種レベルでの分類が基本となっています。従って、対象微生物の属・種名を適切に同定することが、安全性評価を実施するための最初のステップであると言えます。
細菌の「種」の同定には、 DNA-DNA Hybridization (DDH)法により定量した2株間のゲノム配列の類似度 (= DNA-DNA交雑率)が基準とされており、一般的に類似度が70%以上あれば同種と判定されます(Wayne et al., 1987, IJSB 37: 463)。しかし、DDHは操作が煩雑で、実験操作 (温度、DNAの精製度、DNAの断片化度合いなど) に起因する誤差が非常に出やすく、実験者の熟練を要するため、容易には行えません。このことから、簡易的に菌種を同定する場合には、16S rRNA遺伝子配列の類似度が利用されています。ほぼすべての既知基準株について、16S rRNA遺伝子配列がデータベース化されており、対象菌株から取得した16S rRNA遺伝子配列をデータベースと比較することで容易に同定することが可能となっています。ただし、 16S rRNA遺伝子配列で明確に区別できない分類群については、複数のハウスキーピング遺伝子を用いた Multilocus sequence analysis (MLSA) 法による菌種の同定が推奨されています。また、近年では、全ゲノム配列の類似度から菌種の同定を行う、Average Nucleotide Identity (ANI)法も使用されて来ています。
バイオレメディエーション等の環境微生物利用分野においては、浄化剤等が生態系に与える影響を評価する上で、環境中における微生物叢解析を実施する場合があります。近年では、新型シーケンサーと呼ばれる解析装置の登場により、比較的安価で大量のシーケンスが得られる状況になって来ました。このため、環境中の網羅的な菌叢解析に新型シーケンサーを活用する事例が増えてきています。
微生物の分子系統解析 ~Multilocus sequence analysis(MLSA)について~
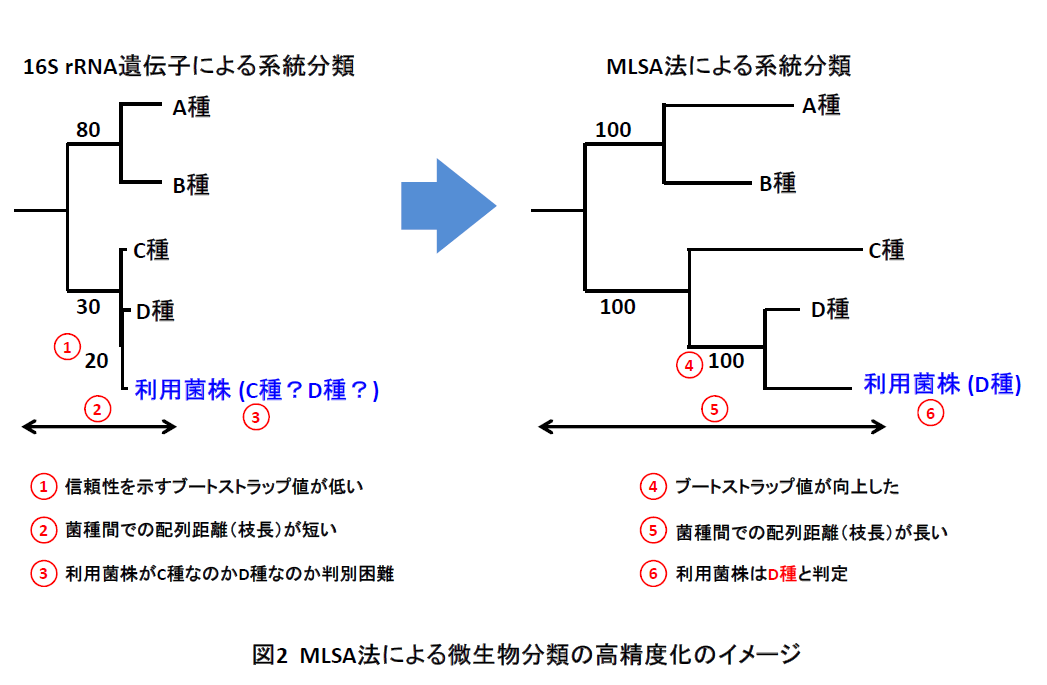
微生物の有用性や安全性の判断には、利用微生物の属種名を決定する系統学的な同定を行う必要があります。このため、微生物の研究開発、医療分野における臨床検査、食品製造における品質管理等の様々な局面において、微生物の同定が行われています。バクテリアの分類においては、 16S rRNA遺伝子配列に基づいた系統解析法が行われていますが、別種間でも配列が完全に一致するケースが散見されるようになり、その解像度の低さが問題視されつつあります。 そこで、国際原核生物分類命名委員会(International Committee on Systematics of Prokaryotes, ICSP)は、16S rRNA遺伝子配列が98.5%以上類似していて区別できない場合、 ハウスキーピング遺伝子*1を用いたMultilocus sequence analysis (MLSA) による菌種の同定を推奨しています(Stackebrandt et al. 2002, IJSEM 52:1043)。 MLSAは図1のように、複数のハウスキーピング遺伝子(5種類以上推奨)の塩基配列またはアミノ酸配列を連結して解析する方法であり、 16S rRNA遺伝子による系統解析と比べ、高精度な解析を行うことができます(図2)。ただし、分類群によってMLSA法に用いられるハウスキーピング遺伝子の組み合わせが異なるため、それぞれの分類群に適したMLSA法の開発が望まれています。
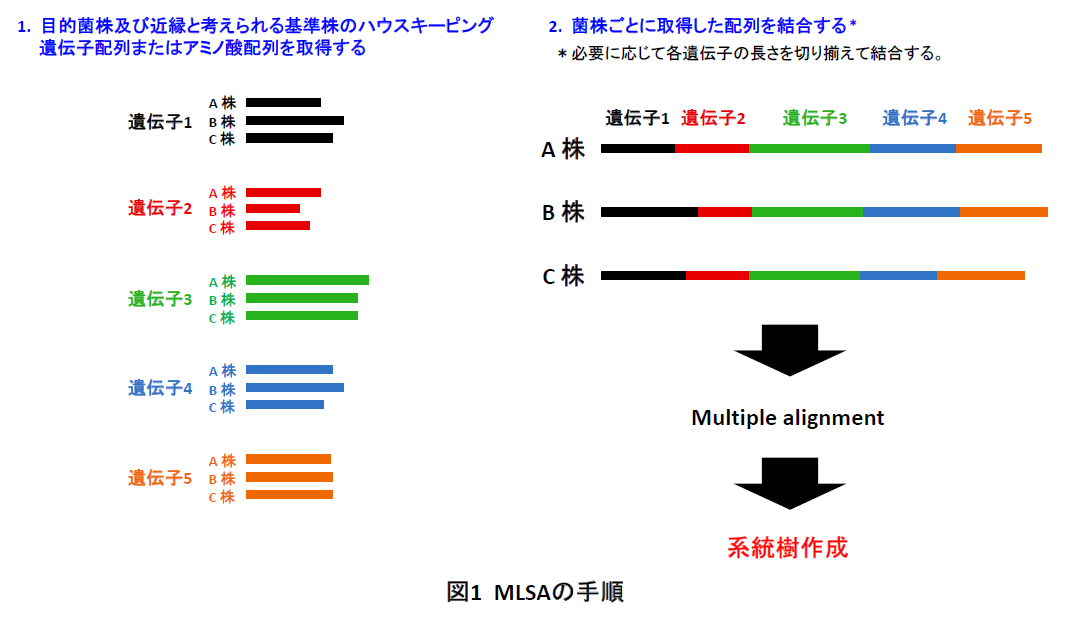
- *1 ハウスキーピング遺伝子:
- 細胞の生育に必須であるため、全細胞で共通に保存されていると考えられる遺伝子のこと。
菌種の同定法 ~ Average Nucleotide Identity (ANI)について~
急速な細菌ゲノム情報の蓄積を背景として、菌株間のゲノム配列の類似度をコンピューター上で計算し、簡便・迅速に菌種の同定を行う、Average Nucleotide Identity (ANI)法がDNA-DNA Hybridization (DDH)法にかわる方法として評価されてきています(Goris et al. 2007, IJSEM 57: 81; Richter and Rosselló-Móra 2009, PNAS 10: 105; 図1)。DDHにおける菌種判定の閾値である類似度70%は、ANIにおいて95-96%に相当すると報告されています。このことから、近年では、DDHの代替としてANIに基づく新種の提案が行われてきています(Baek et al. 2015, IJSEM 65: 504; Shahraki et al. 2015, IJSEM 65: 504)。ANI法はDDHと異なり、実験環境や実験者の熟練度などの影響を受けず、ゲノム配列があれば誰が行っても同じ結果を得られます。また、比較したい菌株のゲノム配列が公開されていれば、そのゲノム配列をANIに使用できること、ANIを行うためのWebサイトや無料のソフトウェアが公開されていることから(Figueras et al. 2014, Genome Announc. 2: e00927-14)、対象としている菌株のゲノム配列さえあれば誰でも行うことができます。ただ、この方法はゲノム情報の蓄積が進んだ分類群では容易に利用できますが、ゲノム情報がない分類群では比較対象菌株も含めてゲノム配列を自ら決定する必要があります。今後、より多くの分類群でゲノム情報の蓄積が進むことで、普及してゆくと考えられます。
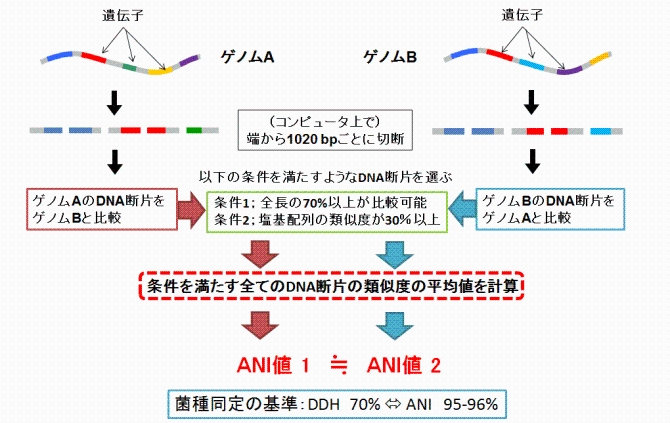
図 ANIの原理
生態系影響評価手法
微生物はあらゆる環境中に存在し、生産者あるいは分解者として物質代謝に大きく関与しています。 その生態を把握するためには、環境中の微生物を検出し、性質を調べていくことが必要ですが、環境に存在する微生物の多く(99%以上)は培養できないことが知られています。 従って、培養法のみでその生態系を評価することは難しいため、近年では培養に依存しない分子生物学的な手法を用いた解析が主流になっています。 また、リボソーマルRNA遺伝子(原核生物;16S rRNA、真核生物;18S rRNA)を標的にした網羅的な微生物叢解析について、代表的な解析手法を以下に示します。
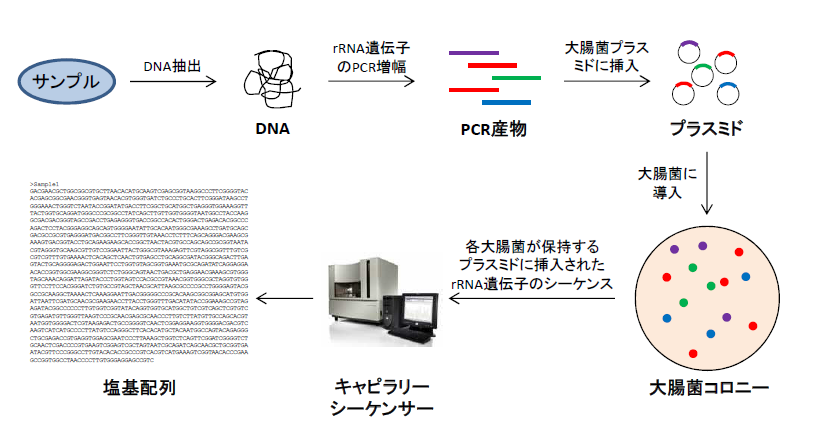
1)クローンライブラリー解析
PCR 産物を大腸菌にクローニングし、各クローンのDNA 塩基配列解析を行い、近縁種を推定する手法である。 クローンライブラリー解析は、長い塩基配列長を対象とすることができるので、より詳細な微生物種の把握に有効である。 反対に、環境中に優占する微生物種の把握のためには、ある程度のクローン数を解析する必要がある。
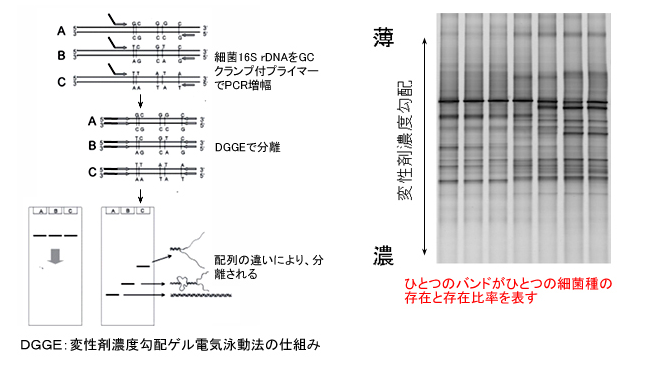
2)DGGE法 (Denaturing Gradient Gel Electrophoresis 変性剤濃度勾配ゲル電気泳動法)
片側のプライマーにGCクランプを付加したプライマーセットでPCR増幅し、変性剤濃度勾配ゲルで電気泳動する。 増幅された二本鎖DNAは分子量の違いだけではなく、変性のしやすさの違いによって移動度が異なることを利用して、微生物叢をバンドパターンとして可視化することができる。 また、ゲルから DGGEバンドを切り出してDNA塩基配列を決定し、得られたDNA塩基配列に基づき系統解析をすることも可能である。
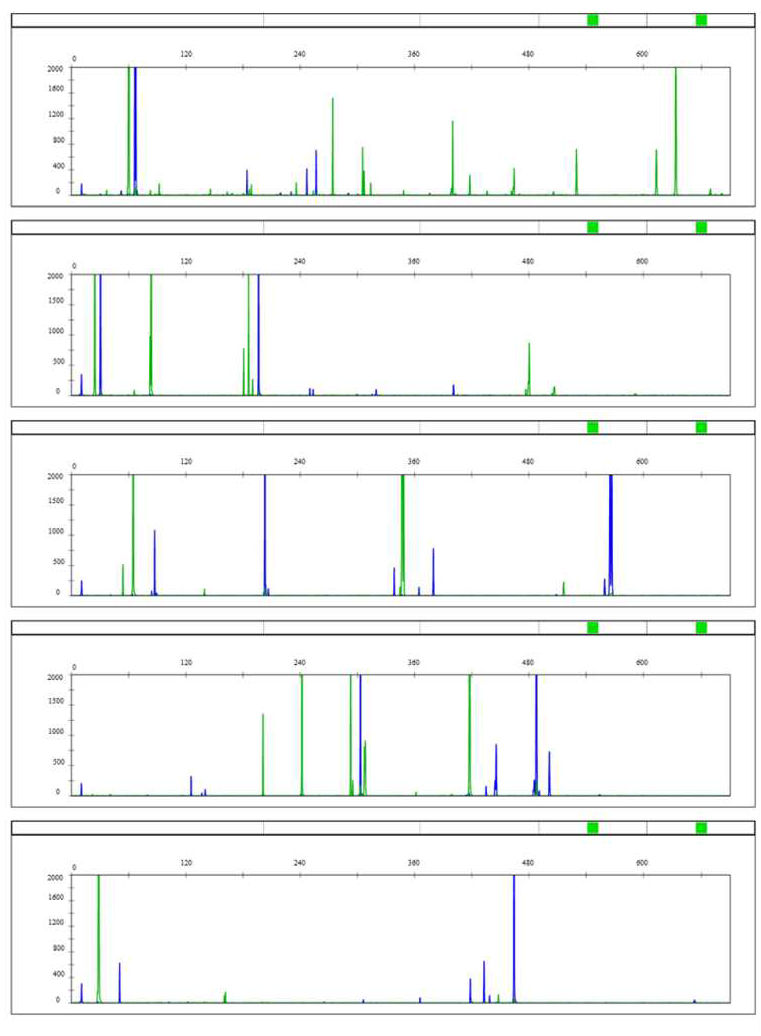
3)T-RFLP法 RFLP(Terminal Restriction Fragment Length Polymorphism; 末端標識制限酵素断片多型分析)法
T-RFLP法は、末端蛍光標識したプライマーセットで鋳型DNAをPCR増幅し、制限酵素による消化後、フラグメント解析をする。 DNA塩基配列の違いから制限酵素切断部位が異なることを利用し、検出ピークの強度、位置、数により評価・比較する手法である。
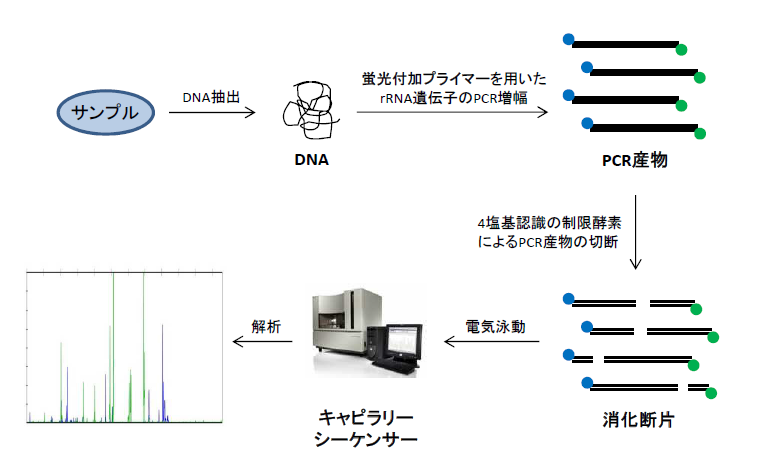
4)新型シーケンサーを用いた アンプリコンシーケンス
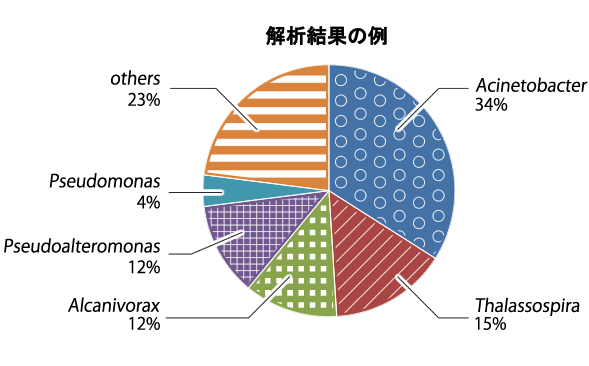
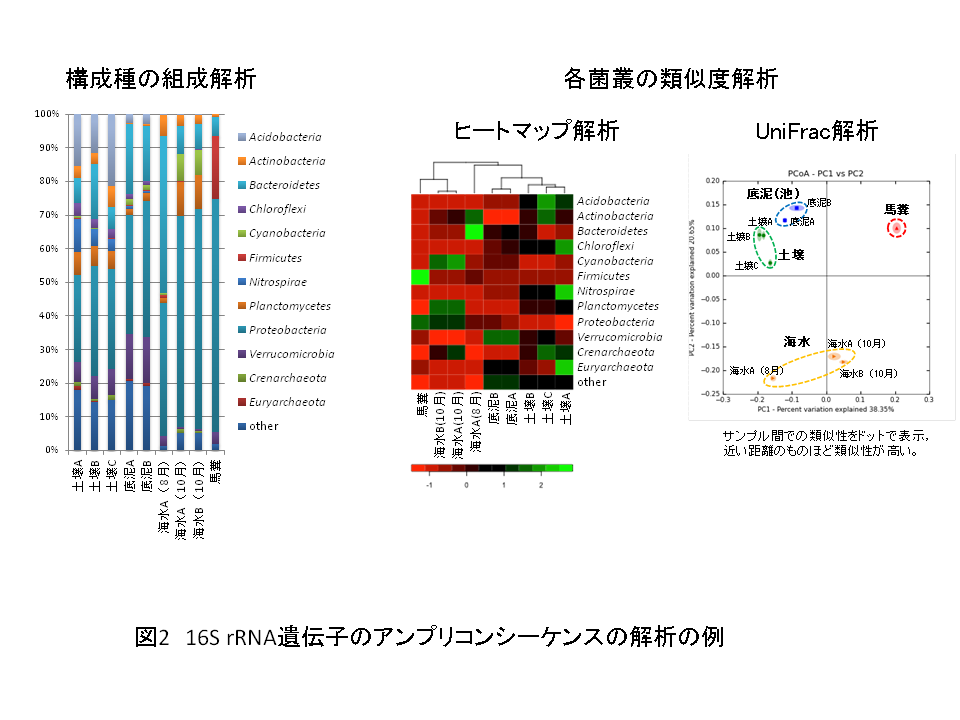
新型シーケンサーを用いたアンプリコンシーケンスは従来法(クローンライブラリー解析、DGGE、T-RFLP等)では検出できなかった占有率の低い菌種の検出が可能だと考えられています。また、PCRプライマーの一端に"タグ"と呼ばれる配列を付加し、サンプル毎に異なるタグ配列を用いることで、同時に複数サンプルを解析できるので、 多検体、多数の配列解析を行う場合のコストパフォーマンスにも優れています。 このため、NGSの普及にともない、その圧倒的な配列決定能力を環境中微生物の菌叢解析に適用する事例が増えてきています。 NGSを利用した16S rDNAのアンプリコン解析(菌叢解析)では、環境より抽出したDNAからPCR法で16S rRNA遺伝子を増幅した後、NGSを用いて網羅的に配列を決定し、低クオリティリードやキメラ配列の除去を行った後、配列同士をクラスタリングしてOTU (Operational Taxonomic Unit)解析を行います。OTUとは、ある一定以上の類似性(一般的には96-97%)を持つ配列同士を一つの菌種のように扱うための操作上の分類単位です。従って、OTU数は菌叢を構成する菌種の数を表し、同一のOTUに属するリードの数はその種の相対的な存在量を表していると考えられます。また、各OTUに属するリード数の中から代表的な配列を選び、データベース検索により属種名の同定が可能です。
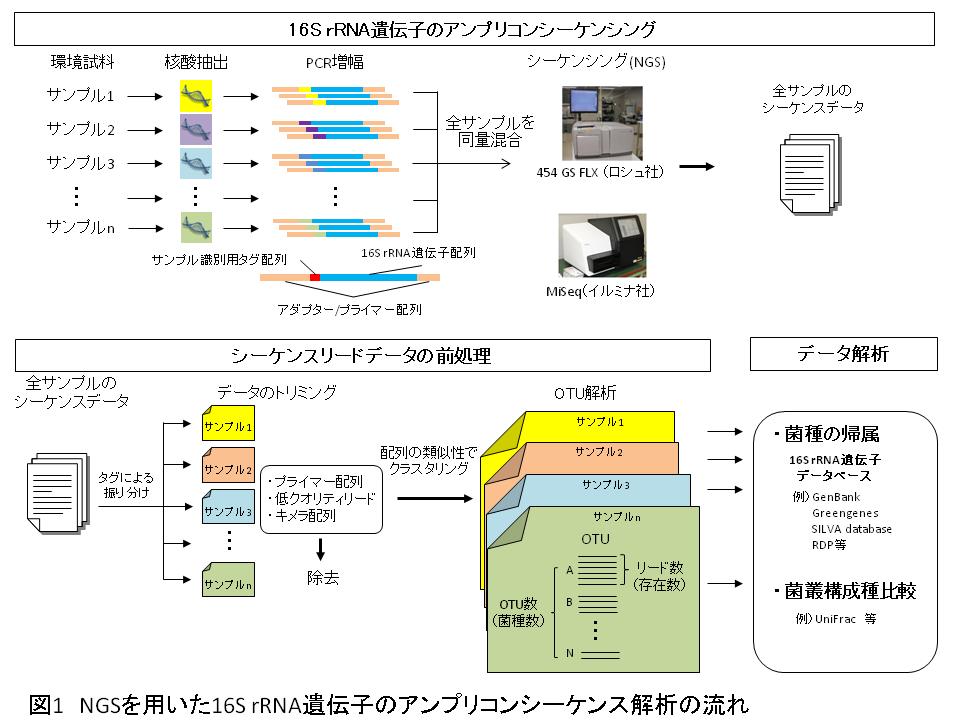
生態系影響評価手法手順書の入手
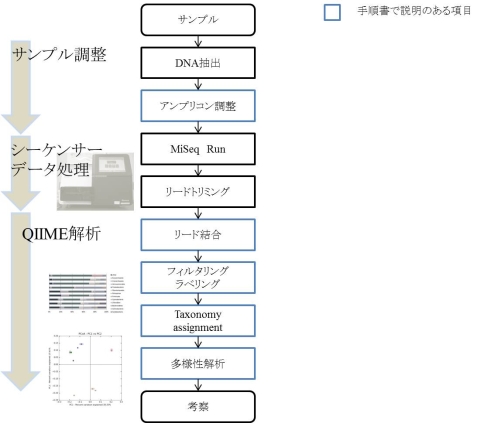
図 環境サンプルからの新型シーケンサーを用いたアンプリコンシーケンスフローチャート
| 様式 | 内容 | 最終更新日 | ダウンロード |
|---|---|---|---|
| バイオレメディエーション における生態系影響評価 手法 |
Illumina社MiSeqシーケンサーを用いた、環境中の微生物生態系解析の実験手法およびパイプラインツール「QIIME」を用いたデータ解析手法の手順書です。 | 2016年3月25日 | 生態系影響評価手法手順書 【PDF:525KB】 |
PDFファイルをご覧いただくためには、Adobe Reader(無償)が必要です。Adobe Readerはダウンロードページ![]() よりダウンロードできます。
よりダウンロードできます。
お問い合わせ
- 独立行政法人製品評価技術基盤機構 バイオテクノロジーセンター バイオデジタル推進課
-
TEL:03-3481-1972
FAX:03-3481-1962
住所:〒151-0066 東京都渋谷区西原2-49-10 地図
お問い合わせフォームへ









